



Publicado
FUNCIONES DE CONTORNO PARA ÁRBOLES COMO MEDIDADE SIMILITUD ENTRE CAMPOS ESCALARES MOLECULARES.UN ESTUDO DE SIMILITUD MOLECULAR
FUNCIONES DE CONTORNO PARA ÁRBOLES COMO MEDIDA DE SIMILITUD ENTRE CAMPOS ESCALARES MOLECULARES. UN ESTUDO DE SIMILITUD MOLECULAR
CONTOUR TREE FUNCTIONS OVER MOLECULAR SCALAR FIELDS REPRESENTATIONS. A MOLECULAR SIMILARITY STUDY
FUNÇÕES DE CONTORNO DE ÁRVORE EM REPRESENTAÇÕES DE CAMPOS ESCALARES MOLECULARES. UM ESTUDO DE SIMILARIDADE MOLECULAR
Julio R. Maza1, Raúl E. Torres1 Edgar E. Daza1,2
1 Universidad Nacional de Colombia, sede Bogotá, Facultad de Ciencias, Departamento de Química, Grupo de Química Teórica. Av Cra 30 45-03- Bogotá D.C., Código Postal 111321, Colombia
2 Universidad Nacional de Colombia, sede Bogotá, Facultad de Ciencias, Departamento de Química, Grupo de Química Teórica. Av Cra 30 45-03- Bogotá D.C., Código Postal 111321, Colombia eedazac@unal.edu.co
Recibido: 20/11/11- Aceptado: 23/12/11
RESUMEN
En este trabajo se implementó una función de contorno de un árbol para establecer medidas de similitud molecular. El estudio se realizó con 73 moléculas orgánicas divididas en 8 grupos funcionales optimizadas con un nivel de teoría DFT//B3LYP/6-31G(d,p) a las cuales se les calculó el Potencial Electrostático Molecular y el Laplaciano de la Densidad Electrónica en una rejilla tridimensional. A partir de los valores de estas propiedades, caracterizando y codificando según su topología, se generaron grafos (árboles) que se compararon a través de la función propuesta. La caracterización y clasificación de las moléculas orgánicas con el Potencial Electrost·tico Molecular muestra una separación correspondiente a moléculas que en su estructura poseen heteroátomos con funciones químicas por lo menos estructuralmente similares, y con el Laplaciano de la Densidad ElectrÛnica se obtuvo como resultado una clasificación acorde con el número de pares de electrones libres asociados a los heteroátomos en las moléculas y a la naturaleza de los átomos que los aportan. Lo anterior evidencia que las funciones de contorno de árbol propuestas en el estudio son una alternativa rápida para clasificar a grosso modo moléculas orgánicas.
Palabras clave: potencial electrostático molecular, Laplaciano de la densidad electrónica, similitud molecular, grafos, clasificación molecular, Taris, funciones de contorno de árboles
ABSTRACT
In this job, Contour Tree Functions were implemented to establish molecular similarity measures. The study was carried by using 73 organic molecules, divided in 8 functional groups and optimized at theory level DFT//B3LYP/6-31G(d,p). The molecular electrostatic potential and the Laplacian of the electron density in a 3D grid for each one were calculated. From the values of these properties, characterizing and encoding the topology, we generated graphs (trees) that are compared with the proposed function. The characterization and classification of the organic molecules with the molecular electrostatic potential show a separation corresponding to molecules that have heteroatoms in their structure with at least similar chemical functions; with the Laplacian of the electron density we achieved a classification according to the number of free pairs of electrons associated to the heteroatoms in the molecules and to the nature provided by the heteroatoms. This is evidence that Contour Tree Functions proposed in this study are a quick alternative to broadly classify organic molecules.
Key words: Molecular electrostatic potential, Laplacian of the electron density, molecular similarity, graphs, molecular classification, TARIS, contour tree functions
RESUMO
Neste trabalho, funções de contorno de árvore foram implementadas para estabelecer medidas de similaridade molecular. O estudo foi conduzido com 73 moléculas orgânicas, divididas em 8 grupos funcionais, otimizadas com nível de teoria DFT//B3LYP/6-31G (d,p) para que eles calculassem o potencial eletrostático molecular e o Laplaciano da densidade de elétrons em uma grade tridimensional. A partir dos valores dessas propriedades, caracterização e codificação da topologia, geramos gráficos (árvores) que são comparados com a função proposta. A caracterização e classificação de moléculas orgânicas com potencial eletrostático molecular mostra uma separação correspondente às moléculas que possuem heteroátomos em sua estrutura com funções químicas, pelo menos, estruturalmente semelhantes. Com o Laplaciano da densidade de elétrons foi obtido como resultado consistente com a classificação do número de pares de elétrons livres associados com heteroátomos nas moléculas e a natureza dos átomos que contribuem. Essa é uma evidência de que as funções de contorno de árvore proposto no estudo são uma alternativa rápida para classificar, grosso modo, moléculas orgânicas.
Palavras-chave: potencial eletrostático molecular, laplaciano da densidade de elétrons, similaridade molecular, gráficos, classificação molecular, TARIS, funções de contorno de árvore.
INTRODUCCIÓN
El principio fundamental de estudios QSAR establece que compuestos estructuralmente similares tienden a exhibir comportamientos similares (1-3), por tanto es imperativo disponer de métodos para cuantificar la similitud molecular. La necesidad de establecer una comparación cuantitativa entre moléculas no es un campo nuevo en química (4), por ejemplo, en la química médica se recurre al concepto de bioisosterismo, según el cual se pueden intercambiar subestructuras similares entre compuestos manteniendo un cierto grado de actividad biológica (5). Este tipo de conceptos ha sido aprovechado en especial por la industria farmacéutica, que ve en los métodos teóricos de similitud molecular una gran alternativa para el diseño de fármacos (6, 7). De manera que cabe preguntarse en qué sentido se considera esta similitud.
Una dificultad inherente al desarrollo de métodos teóricos de similitud molecular radica en que tanto el concepto de estructura molecularî como el de ìsimilitud no están plenamente definidos. En general, la comparación de moléculas para establecer similitud molecular busca patrones comunes en algún tipo de representación molecular referidos a un objetivo predeterminado, por tanto, diferentes representaciones conducirán, en principio, a diferentes relaciones, inclusive una misma representación puede dar lugar a diferentes clasificaciones si se consideran distintos objetivos. A pesar de estas dificultades, la investigación sobre medidas de similitud continúa (8, 9).
En este trabajo empleamos como representación molecular dos propiedades: el Potencial Electrostático Molecular (PEM) y el Laplaciano de la Densidad Electrónica Molecular (LDEM), que son campos escalares asociados a una geometría molecular dada. A diferencia de la mayoría de las metodologías desarrolladas para comparar campos escalares moleculares tales como CoMFA (Comparative Molecular Field Analysis) (10) y QMSM (Quantum Molecular Similarity Measures) (11, 12), las cuales requieren de un alineamiento previo de las geometrías nucleares, nuestra propuesta no demanda ese proceso. Esta particularidad es una gran ventaja, ya que alinear esqueletos moleculares es un problema complejo que conlleva un alto costo computacional que crece con la cantidad de moléculas por alinear y con la complejidad de sus esqueletos moleculares (13-15). El método propuesto parte de los valores del campo alrededor de la molécula para establecer las isosuperficies asociadas a los mínimos locales del mismo, y monitorear la aparición o fusión de ellas para establecer una correspondencia con un grafo que condensa las principales características topológicas del campo. En este caso particular, el tipo de grafo resultante es un árbol, cuyos nodos pueden ser ponderados, por ejemplo, con el valor del área de la superficie correspondiente, con el fin de dar mayor flexibilidad a la representación del campo (13, 16).
Aquí presentamos una extensión a la metodología desarrollada para comparar moléculas usando el PEM (13, 16) para incluir otra propiedad, el Laplaciano de la Densidad Electrónica Molecular, como otro campo escalar para el estudio de similitud molecular. También presentamos una forma alternativa para comparar los árboles con que codificamos los campos escalares, ahora mediante una función de contorno del árbol (17) en lugar de la costosa métrica de edición que se ha empleado con anterioridad. Las funciones de contorno se han empleado en otras ciencias (18), por ejemplo, en medicina, para el análisis de imágenes médicas.
La nueva medida de similitud se empleó para clasificar, a partir del PEM, un conjunto representativo de los distintos grupos funcionales de la química ya que son muchas las interacciones a nivel intermolecular que están gobernadas por esta propiedad (19-21). Además, se estudió el mismo conjunto de moléculas, ahora representado mediante el LDEM, que permite establecer dónde se acumula la carga electrónica, y a partir de esta información, dar cuenta de los pares de electrones libres del modelo de Lewis (22). En ambos casos, los resultados evidencian que las funciones de contorno de un árbol son una alternativa rápida para comparar grosso modo el conjunto de moléculas representadas por estos campos escalares que conllevan a clasificaciones promisorias.
MATERIALES Y MÉTODOS
Moléculas de estudio
Se escogieron 73 moléculas orgánicas representativas de 8 grupos funcionales (ver Tabla 1).
Propiedades moleculares
El Potencial Electrostático Molecular PEM puede determinarse experimentalmente (22) o ser calculado en un punto del espacio (r) a partir de la densidad de carga (r) estimada mediante métodos de la química cuántica computacional para una disposición de los núcleos a partir de:

El Laplaciano de la Densidad Electrónica Molecular LDEM es un escalar definido como la traza de la matriz de segundas derivadas de la función de densidad electrónica:

Es de resaltar que en el presente trabajo se aprovechan por primera vez las características topológicas del LDEM codificadas como un grafo para su posterior comparación. La topología del LDEM había sido usada por Bader en su teoría de Átomos en Moléculas (23) para la caracterización local de puntos críticos o determinar órdenes de enlace (24).
El cálculo de estas dos propiedades se realizó para todas las moléculas en sus geometrías de equilibrio a partir de densidades calculadas utilizando la teoría de los funcionales de la densidad (DFT) con el funcional híbrido B3LYP y una base electrónica 6-31G (d,p), empleando el programa Gaussian 98 (25), sobre una rejilla tridimensional con una densidad aproximada de 90puntos/Bohr3.
Los campos escalares de cada una de las moléculas se transformaron en un árbol por molécula, siguiendo el procedimiento descrito en (13, 16), según se esquematiza en la Figura 1. Para ello se empleó el programa TARIS con los siguientes parámetros para la búsqueda por niveles: i) para el Potencial Electrostático Molecular, inicio de la búsqueda 0.10 u.a. y fin de la búsqueda 0.04 u.a., ii) para el Laplaciano de la Densidad Electrónica Molecular: inicio de la búsqueda 1.00 u.a. y fin de la búsqueda 0.16 u.a.; para ambos campos escalares, el tamaño del paso de la búsqueda fue de 0.01 u.a.
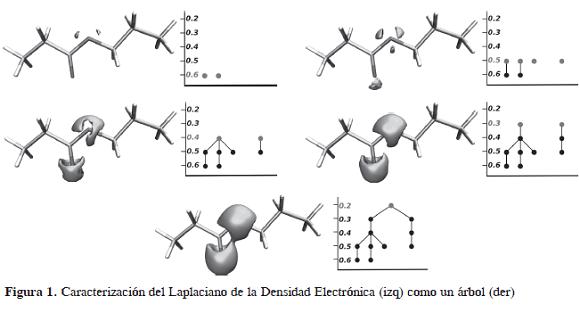
Con las propiedades codificadas como árboles, la etapa siguiente para establecer similitudes moleculares es la comparación de los árboles; en el presente trabajo propusimos calcular las funciones de contorno de los árboles como una alternativa para la comparación de árboles, que anteriormente se realizaba con la métrica de edición de árboles (26).
Funciones de contorno de Árbol
El primer paso para definir las funciones de contorno es implementar un algoritmo de búsqueda o recorrido a través de los vértices, guardando el valor de cada nodo visitado. Usamos el algoritmo de búsqueda conocido como Depth-first (27); una manera gráfica de representar el algoritmo se muestra en la Figura 2. En este árbol de ejemplo, los vértices están ponderados con el valor del nivel donde aparecen, así el nodo raíz tiene un valor de cero, y a medida que aumentamos en profundidad el valor del nivel se incrementa en uno. Comenzamos la búsqueda por la izquierda hacia el nodo más profundo y vamos registrando cada nodo encontrado, como se indica sistemáticamente en la Figura 2, y generamos un contorno del árbol mediante el recorrido.
Las funciones de contorno no se pueden comparar punto a punto, pues no hay correspondencia entre los nodos de los árboles, por lo que propusimos el área bajo las funciones como magnitud para comparar entre distintas funciones de contorno. Para esto último empleamos un método de integración numérica basado en la regla trapezoidal. El valor absoluto de la diferencia entre dos de estas áreas corresponde a la disimilitud entre los correspondientes árboles, es decir, entre los campos asociados a un par de moléculas. De esta forma, una vez determinadas todas las distancias, construimos las matrices de distancia para las moléculas del estudio. Establecimos una medida de similitud molecular, generando la matriz de similitud normalizando las distancias con respecto a la distancia máxima obtenida.


Esta forma de comparar árboles se implementó computacionalmente como un nuevo módulo dentro del programa TARIS, de manera que ahora se cuenta con más de una forma para evaluar la similitud de campos escalares moleculares (Laplaciano de la Densidad Electrónica y Potencial Electrostático Molecular) representados como grafos.
RESULTADOS
A partir de las matrices de distancia es posible establecer una clasificación de las moléculas según su similitud o diferencia. Para ello empleamos una técnica de agrupamiento bien conocida, como lo es el agrupamiento jerárquico promedio no ponderado (28). Los resultados de los agrupamientos obtenidos para cada una de las dos propiedades se presentan en forma de dendrogramas. Mostramos los resultados divididos en dos, lo que corresponde con los dos campos escalares estudiados, el PEM y el LDEM.
PEM: Al leer el dendrograma correspondiente al PEM (Figura 3), observamos dos grandes ramas: una primera, en la parte superior, que involucra aquellas moléculas que no poseen grupo carbonilo en su función, rama conformada por los grupos A, B y C, con la excepción del grupo X cuyo comportamiento se discutirá más abajo. Una segunda rama conformada por los grupos D, E y F que corresponde a moléculas en las cuales hay, al menos, un oxígeno carbonílico.

Con base en la formación de los grupos A, B, C y X, también se observa que hay una buena separación por grupos funcionales, así: el grupo A, conformado mayormente por las aminas (AMN) y un par de éteres el ETR03 y ETR09. El grupo B, constituidos exclusivamente por éteres (ETR); se debe resaltar que para los éteres, el tamaño de las moléculas es un factor importante en en su clasificación, pues estos, con una estructura molecular pequeña (ETR01-ETR02, ETR08), se encuentran separados de los éteres con un mayor número de carbonos (ETR05-ETR07, ETR04). El grupo C está formado exclusivamente por la serie de alcoholes (ALC). El grupo X presenta una mezcla de moléculas correspondientes a aldehídos (ALD) y ácidos carboxílicos (ACD) pequeños: los ácidos ACD01, ACD02, ACD03, ACD04 y el aldehído ALD01. Para estas moléculas, el PEM queda determinado por el oxígeno y no alcanza a afectarse de forma suficiente por los demás átomos de la cadena.
En el segunda rama no se logra una separación total por grupos funcionales, sin embargo, el dendrograma permite establecer un patrón: el grupo D, conformado por una mezcla de ácidos carboxílicos (ACD) y aldehídos (ALD), tiene los oxígenos (COOH) y (COH) expuestos, en contraste con las moléculas en el grupo E, constituido por los ésteres (EST) y cetonas (CET), cuyas moléculas poseen una cadena carbonada R en reemplazo del hidrógeno. Esta diferencia se refleja en el comportamiento del PEM que da lugar a esta clasificación.
Por último, se tiene el grupo F, conformado por las amidas (AMD); internamente hay una separación que responde a la estructura de las moléculas; así podemos notar un grupo formado por las moléculas AMD04-AMD05, AMD03 cuyas estructuras moleculares presentan la función amida sobre una cadena carbonada mayor (superior a tres átomos de carbono), mientras que en la otra agrupación, dada por las moléculas AMD01-AMD06, AMD02, que tienen en la cadena principal solo uno o dos átomos de carbono.
En un estudio previo realizado en nuestro grupo, se utilizó la métrica de edición de árboles como medida de disimilitud entre árboles, de la que se dice es una de las mejores medidas (29). Los resultados obtenidos mostraron agrupamientos que concuerdan con la clasificación más básica de las moléculas orgánicas, los grupos funcionales, con algunas excepciones de moléculas de tamaño menor en comparación con sus series homólogas (ver Figura 4).
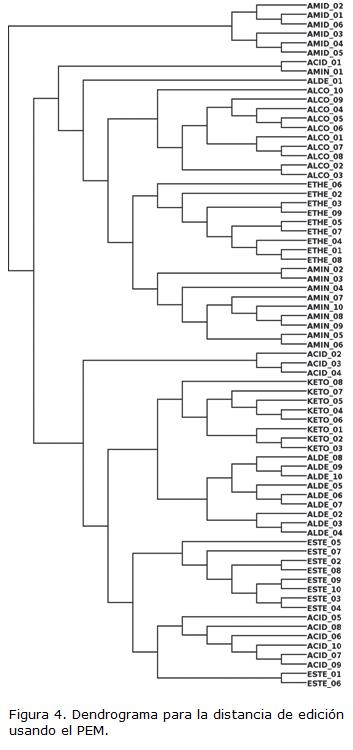
Si bien hay diferencias entre los dos dendrogramas, y, por tanto, las clasificaciones no son exactamente las mismas, hay un buen grado de correlación entre las dos matrices de distancia, una correlación de 0,9606, por lo que en términos generales puede decirse que, grosso modo, esta nueva medida de similitud produce los mismos resultados; pero si se tiene en cuenta que la métrica de edición de árboles es un proceso computacionalmente muy costoso, y que, por el contrario, las áreas bajo las funciones de contorno son muy fáciles de calcular, podemos afirmar que las últimas son una alternativa en la comparación de moléculas (Para el ejemplo actual de comparación de áreas, el tiempo fue de 0,00175 segundos). El uso de esta medida de similitud resulta aconsejable como primera opción cuando el número de árboles por comparar es grande y luego, si se quiere, refinar la clasificación dentro de subgrupos especiales empleando la métrica de edición.
LDEM: El Laplaciano de la Densidad Electrónica es una propiedad diferente al PEM y por lo tanto los resultados obtenidos son también diferentes. En este caso, al utilizar la misma técnica de agrupamiento sobre la matriz de distancias encontramos la siguiente clasificación (Figura 5).
En el dendrograma correspondiente al LDEM se establecen cuatro ramas bien definidas. Cada una de ellas corresponde a moléculas con un mismo número y tipo de heteroátomos. En el grupo denominado A están las moléculas con las funciones químicas ácido carboxílico y ésteres, es decir, moléculas con dos heteroátomos iguales: un oxígeno carboxílico, C=O, y el otro unido a un hidrógeno, en el caso del ácido, o a una cadena carbonada, en el caso de los ésteres, O-H(R). En el siguiente conglomerado B encontramos a las amidas, función con dos heteroátomos diferentes: un oxígeno carbonílico y un nitrógeno. El grupo C reúne los alcoholes, éteres, aldehidos y cetonas, cada una de estas funciones químicas tiene un solo heteroátomo. Por último, en el dendrograma aparece el grupo D que reúne a las aminas, también con un solo heteroátomo.

El LDEM mide la concentración de la densidad electrónica (23, 24), que justifica su uso para la detección de la localización de pares de electrones. Nuestros resultados muestran que, en efecto, su uso permite una clasificación de las moléculas estudiadas discriminándolas por el número de pares de electrones libres que posean. Así, el grupo A con 4 pares de electrones libres (2 de cada oxígeno); el grupo B las amidas con 3 pares de electrones libres (2 del oxígeno y 1 del nitrógeno); el grupo C óalcoholes, éteres, aldehidos y cetonas con 2 pares de electrones libres correspondientes al oxígeno, y el grupo D, las aminas con 1 par de electrones libres.
CONCLUSIONES
El estudio de similitud molecular realizado sobre el conjunto de 73 moléculas orgánicas distribuidas en ocho grupos funcionales, usando el PEM, dio como resultado agrupamientos por grupo funcional similares a los obtenidos mediante la métrica de edición.
El estudio de similitud molecular, usando el LDEM, arrojó una clasificación acorde con el número pares de electrones libres asociados a los heteroátomos en las moléculas, y a la naturaleza de los átomos que los aportan.
El uso de las áreas bajo las funciones de contorno son una gran alternativa para una rápida comparación de árboles con raíz ponderados, con un costo computacional 20 veces menor, pero con la pérdida de información correspondiente a la topología de los campos escalares calculados.
REFERENCIAS BIBLIOGRáFICAS
1. Thorner, D.; Wild, D.; Willett, P.;Wright, P.M. Calculation of structural similarity by the alignment of molecular electrostatic potentials. En: 3D QSAR in Drug Design. Netherlands : Springer. 2002: 301-320.
2. Bender, A.Studies on Molecular Similarity. PhD Thesis, Darwin College. 2006.
3. Johnson, M.A.; Maggiora, G.M. Concepts and applications of molecular similarity. New York. Wiley Interscience Publication. 1990.
4. Matta, C.F.; Alya, A.; Donald, F. The bioisosteric similarity of the tetrazole and carboxylate anions: Clues from the topologies of the electrostatic potential and of the electron density. Eur. J. Med. Chem. 2010. 45: 1868-1872.
5. Martin, Y.; Kofron, J.; Traphagen, L. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002. 45: 4350-4358.
6. Liljefors, T. 3D QSAR in drug design: Ligand-Protein interactions and molecular similarity. Vol 2. Kluwer Academic Publishers. 2002: 3-17.
7. Sheridan, R.P. The most common chemical replacements in drug-like compounds. J. Chem. Inf. Comput. Sci. 2002. 42: 103-108.
8. Garcia-Domenech, R.; Galvez, J.; Pogliani, L. Some new trends in chemical graph theory. Chem. Rev. 2008. 108: 1127-1169.
9. Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998. 38: 983-996.
10. Cramer, R.D.; Patterson, D.; Bunce, J. Comparative molecular-field analysis (Comfa): Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988. 110: 5959-5967.
11. Carbo, R.; Leyda, L.; Arnau, M. An electron density measure of the similarity between two compounds. Int. J. Quant. Chem. 1980. 17: 1185-1189.
12. Carbo-Dorca, R.; Besalu, E. A general survey of molecular quantum similarity. J. Mol. Struct. 1998. 451: 11-23.
13. Marin, R.; Aguirre, N.; Daza, E. Graph theoretical similarity approach to compare molecular electrostatic potentials. J. Chem. Inf. Model. 2008. 48: 109-118.
14. Pastor, M.; Cruciani, G.; McLay, I.; Pickett, S.; Clementi, S. GRid-INdependent Descriptors (GRIND): a novel class of alignment-independent three-dimensional molecular descriptors. J. Med. Chem. 2000. 43: 3233-3243.
15. Wagener, M.; Sadowski, J.; Gasteiger, J. Autocorrelation of molecular surface properties for modeling corticosteroid binding globulin and cytosolic ah receptor activity by neural networks. J. Am. Chem. Soc. 1995. 117: 7769-7775.
16. Daza, E.; Maza, J.; Torres, R. Molecular electrostatic potential as a graph. CCADD. Topology and Chemical Connectivity. (In Press)
17. Bajaj, C.; Pascucci, V.; Schikore, D.R. The Contour spectrum. Proceedings of IEEE Visualization. 1997. 1: 167-173
18. Carr, H.; Snoeyink, J.; Axen, U. Computing contour tree in all dimensions. SIAM Symposium on Discrete Algorithms. 2000. 11ACM: 928-926.
19. Sadlej-Sosnowska, N. Molecular similarity based on atomic electrostatic potential. J. Phys. Chem. A. 2007. 111: 1113411140.
20. Roy, D.K.; Balanarayan, P.; Gadre, S.R. Signatures of molecular recognition from the topography of electrostatic potential. J. Chem. Sci. 2009. 121: 815-821.
21. Bagdassarian, C.K.; Schramm, V.; Schwartz, S. Molecular electrostatic potential analysis for enzymatic substrates, competitive inhibitors, and transition-state inhibitors. J. Am. Chem. Soc. 1996. 118: 8825-8836.
22. Bader, R.F.W. Atoms in molecules: A quantum theory. Oxford University Press. 1999.
23. Murray, J.S.; Sen, K.D. Molecular electrostatic potentials: concepts and applications. Elsevier Science. 1996.
24. Popelier, P.L. Atoms in molecules: An introduction. Prentice Hall. 2000.
25. Gaussian 98, Revision A.11, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman et. al., Gaussian Inc., Wallingford CT, 1998.
26. Bille, P. Tree Edit Distance, Alignment Distance and Inclusion. IT Univ. of Copenhagen. 2003.
27. Nayak, A.; Stojmenovic, I. Handbook of Applied Algorithms. Willey-InterScience. 2008.
28. Huson, D.H.; Richter, D.C. et al. Dendroscope-An interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007. 8:460.
29. Bunke, H.; Riesen, K. Graph Edit Distance: Optimal and Suboptimal Algorithms with Applications. En: Analysis of complex networks, from biology to linguistics. WILEY-VCH Verlag GmbH & Co. 2009: 113-143.
Cómo citar
IEEE
ACM
ACS
APA
ABNT
Chicago
Harvard
MLA
Turabian
Vancouver
Descargar cita
Visitas a la página del resumen del artículo
Descargas
Licencia
Los autores/as conservarán sus derechos de autor y garantizarán a la revista el derecho de primera publicación de su obra, el cuál estará simultáneamente sujeto a la Licencia de reconocimiento de Creative Commons (CC. Atribución 4.0) que permite a terceros compartir la obra siempre que se indique su autor y su primera publicación en esta revista.
Los autores/as podrán adoptar otros acuerdos de licencia no exclusiva de distribución de la versión de la obra publicada (p. ej.: depositarla en un archivo telemático institucional o publicarla en un volumen monográfico) siempre que se indique la publicación inicial en esta revista.
Se permite y recomienda a los autores/as difundir su obra a través de Internet (p. ej.: en archivos telemáticos institucionales o en su página web) antes y durante el proceso de envío, lo cual puede producir intercambios interesantes y aumentar las citas de la obra publicada. (Véase El efecto del acceso abierto).










